

 Download:
Download:




-
Brucellosis represents a globally prevalent zoonotic disease that poses significant public health challenges and causes substantial economic losses in livestock populations (1). The past few decades has witnessed continuous expansion in recognized diversity within theBrucellagenus, with novel strains isolated from marine mammals to ocean fish revealing previously unknown ecological niches (2). These developments present new challenges for both regional and global surveillance and control of brucellosis. However, global species/biovars and genotype diversity atlas ofBrucellaspp. remain unclear. Therefore, this study aims to elucidate the global distribution patterns of species/biovars and genetic diversity amongBrucellastrains to enhance understanding of epidemiological changes and facilitate tailored surveillance and control strategies worldwide.
In this study, 7,212Brucellastrains collected from multiple locus variable-number tandem repeat analysis (MLVA) databases (https://microbesgenotyping.i2bc.paris-saclay.fr/databases) through June 30, 2024, representing isolates collected in 102 countries from 1923 to 2020. Data extraction included species/biovar, isolation location, quantity, host spectrum, panel 1 profiles, MLVA-11 patterns, lineage information, and isolation dates. Data analysis was performed using Excel 2021 software (Microsoft, Redmond, WA, USA). The minimum spanning tree (MST) was constructed using PHYLOVIZ 2.0 (3) online software (https://online2.phyloviz.net/index) to elucidate genetic relationships among strains.
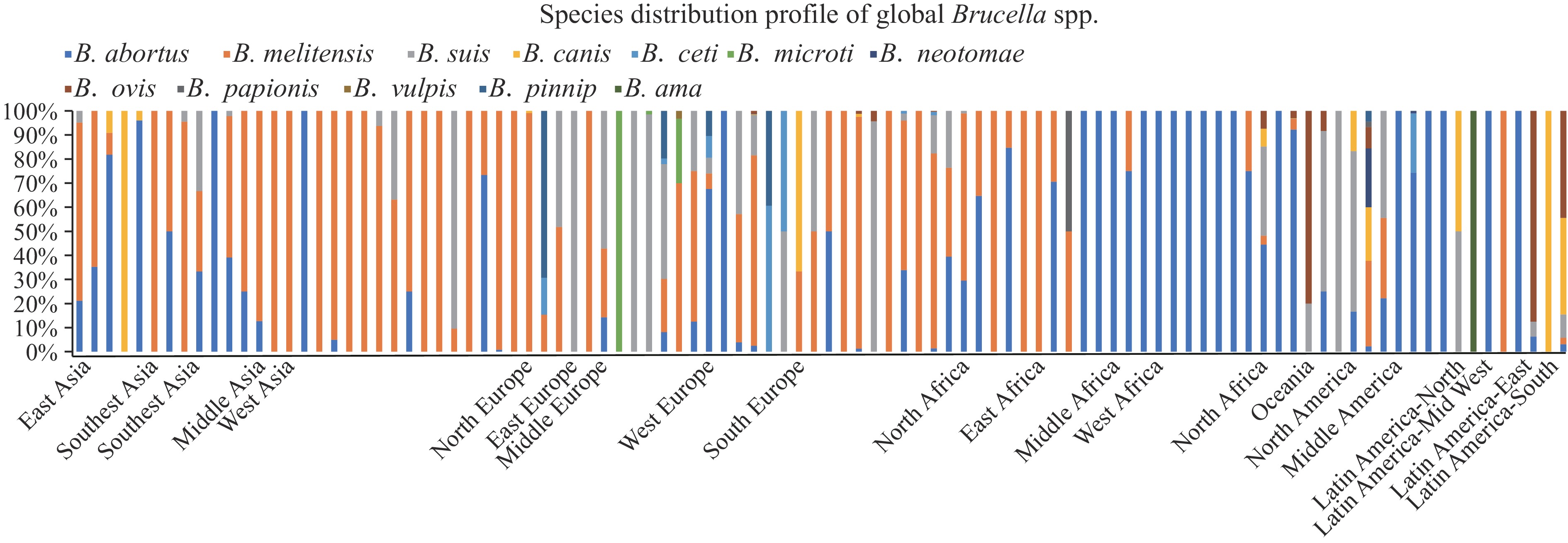
Among the 7,212Brucellastrains analyzed, at least 12 species, 19 biovars, and several atypicalBrucellaspecies were identified (Figure 1).B. abortusstrains distributed across 59 countries (regions) spanning six continents, whileB. melitensisexhibited widespread presence in 64 countries throughout Asia, Europe, and North Africa (Figure 1). Notably, the distribution pattern ofB. melitensiscorrelates strongly with regions reporting high incidence rates of brucellosis in both humans and animals.B. suisstrains were documented in 34 countries across Europe, North America, and Latin America.B. canisdemonstrated a more limited geographic range, primarily concentrated in East Asia and Latin America (Figure 1). Other species showed distinct regional patterns:B. neotomaein North America;B. ovisin Europe, North Africa, Oceania, and Latin America;B. cetipredominantly in West Europe and North America;B. microticoncentrated in Middle Europe;B. papionisrestricted to Tanzania and USA;B. vulpisexclusively in Austria; andB. pinnipprimarily in Europe (Figure 1). These distribution patterns indicate the global predominance ofB. abortusandB. melitensis, while other species exhibit distinct geographic specificities.
 Figure 1.
Figure 1.Territory distribution ofBrucellaspp. Strains.
Note: The number of strains marked with different color scales; green: refers to the continent with no strains found; red: indicates the continent with the most strains.The predominant species wasB. melitensis(n=4,042, 56.05%), followed byB. abortus(n=2,600),B. suis(n=695), andB. canis(n=52) (Supplementary Table S1). The geographic distribution ofB. abortusbiovars showed distinct patterns, for example: biovar 1 was predominantly found in Portugal, South Korea, and Brazil; and biovar 9 in Xinjiang, China. Within theB. melitensispopulation, biovar 3 (n=1,909) was dominated species.B. melitensisbiovars 1 and 3 showed widespread distribution across Asia and Europe, particularly in Asian countries with high brucellosis prevalence.
TheB. suispopulation exhibited distinct biovar distributions, such asB. suisbiovar 1 showed broad geographic distribution across China, the USA, Mexico, France, Zimbabwe, Egypt, and Australia.B. ovis(n=53) was distributed across France, Spain, Brazil, Greece, the USA, Australia, New Zealand, Croatia, and Argentina.B. neotomae(n=13) was confined to Costa Rica and the USA. Among non-classical species, there were 173B. cetiisolates, 61B. pinnipedialisisolates, 13B. microtiisolates, 2B. papionisisolates, and 1B. vulpisisolate.B. cetiwas predominantly found in Scotland, Italy, Spain, Costa Rica, Germany, France, and the UK.B. microtiwas isolated from the Czech Republic and Austria,B. papionisfrom Tanzania and the USA, andB. pinnipedialisfrom Scotland, Norway, United Kingdom, and USA.
Asia exhibits the lowest species diversity, withB. abortusandB. melitensisbeing the only classical species distributed across all Asian countries (Figure 2). Sporadic cases ofB. suishave been documented in China, India, Nepal, Palestine, and the United Arab Emirates, whileB. canishas been reported exclusively in China, Republic of Korea, and Japan. This species distribution profile aligns precisely with the regions reporting the highest human brucellosis burden in Asia. Notably, substantial isolations ofB. melitensishave been recorded in China, Kazakhstan, Kyrgyzstan, Palestine, Qatar, and Turkey - countries that consistently report among the highest global incidence rates of brucellosis.
 Figure 2.
Figure 2.Distribution of areas and composition patterns ofBrucellaspecies populations.
Note: Brucellaspecies are coded with color, and dots on the x-axis indicate the borders of different areas.Europe demonstrates higher species diversity than other continents, with 9 of the 12 known species documented (Figure 2). Three species (B. abortus, B. melitensis,andB. suis) are widely distributed across the continent, particularly in historically high-burden regions.B. suisexhibits a unique continental distribution pattern, with significant presence in Hungary, Germany, Belgium, France, Croatia, Spain, and Portugal, predominantly isolated from swine and wild boar populations. These findings suggest that despite successful control of brucellosis in Europe’s historically endemic areas, continued surveillance remains essential.
Despite Africa being a historically endemic region for brucellosis, comprehensive data onBrucellaspecies and genotypes remain limited due to insufficient surveillance in recent decades.B. abortusstrains are particularly prevalent throughout South and West Africa (Figure 2). In Africa presence of multipleBrucellaspecies, underscoring the need for expanded bacteriological surveillance.
While Oceania maintains brucellosis-free status with only sporadic isolations ofB. abortus,B. suis, andB. ovis(Figure 2), potential public health risks persist through mammalian reservoirs. The Americas exhibit the highestBrucellaspecies diversity globally, with at least 10 documented species, andB. abortusshowing the widest geographic distribution (Figure 2).B. melitensishas been documented in regions with substantial human brucellosis burden, including the USA, Mexico, Peru, and Argentina. The continent harbors the highest concentrations of bothB. neotomae(11 isolates in the USA) andB. ovis(115 isolates in Argentina), while Costa Rica reports the majority ofB. ceticases.
TheB. abortuspopulation demonstrates remarkable host diversity, with isolates from at least 20 different species. Cattle represent the primary host (n=1,567), followed by bison (n=97) and humans (n=80).B. melitensisexhibits even greater host diversity, with isolations from 24 distinct species, predominantly humans (n=2,719), followed by ovines (n=617) and cattle (n=280). TheB. suispopulation spans 15 host species,B. neotomaehas been isolated from both rodents and humans. Host specificity is observed in several species, such asB. papionisin baboons. This extensive host diversity plays a crucial role in maintaining and facilitating the transmission ofBrucellastrains.
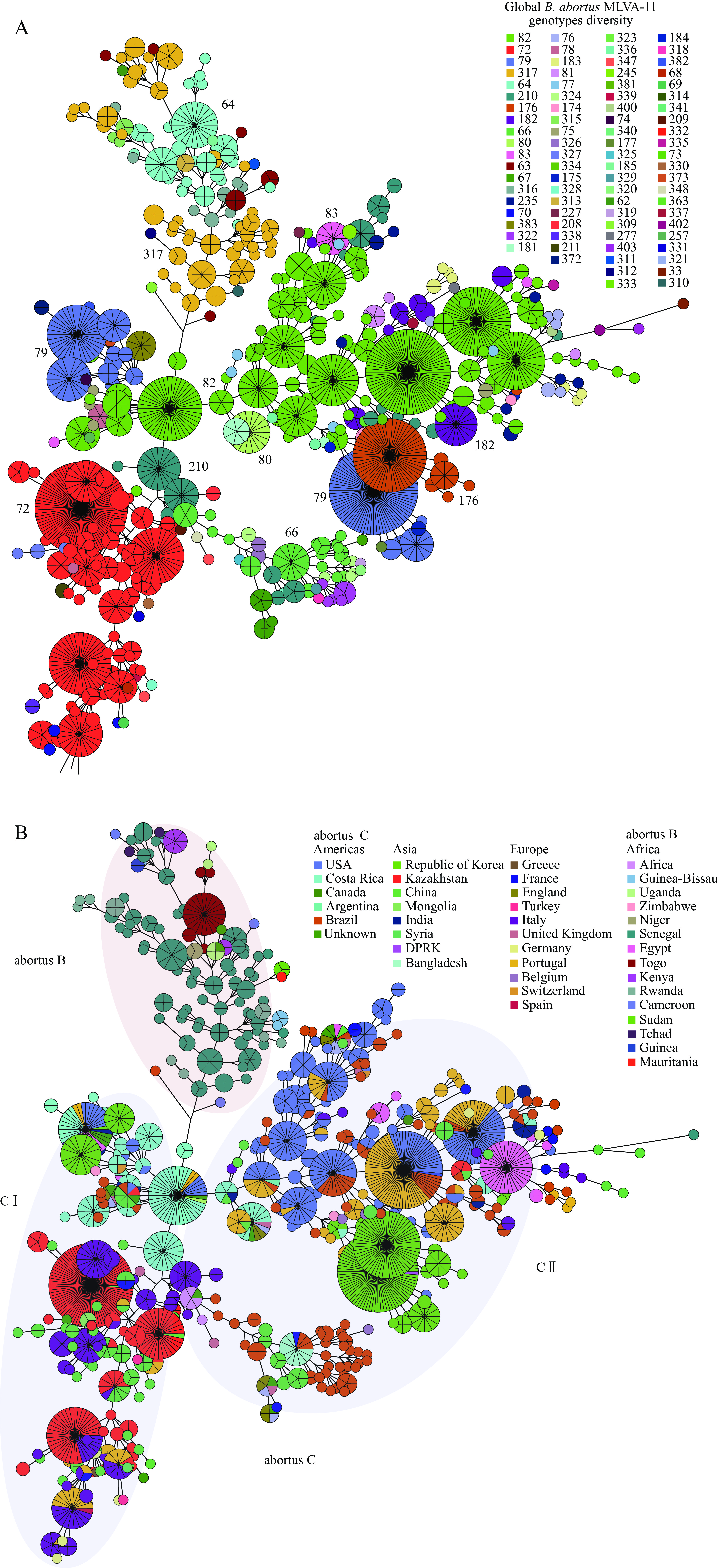
Within theB. abortuspopulation, 83 multiple locus variable-number tandem repeat analysis-11 genotypes (GTs) were identified, with three GTs (82, 72, and 79) emerging as predominant, representing 28.4% (494/1,735), 22.4% (390/1,735), and 10.7% (187/1,735) of the population, respectively (Figure 3A). GT72 exhibited broad geographic distribution across 17 countries, GT82 was detected in 10 countries across 3 continents, GT79 was identified in 9 countries across 4 continents.
 Figure 3.
Figure 3.MLVA-11 genotype diversity (A) and MLVA-16 genetic relationship (B) of globalB. abortusstrains.
Note: (A) Color marks the MLVA-11 genotypes; numbers in the figure indicate the dominant genotypes; (B) Color-coded countries in which strains isolated from all B. abortusstrains were divided into two lineages (abortus A and abortus B), and abortus B were further sorted into C I and C II subgroups.Minimum spanning tree (MST) analysis based on MLVA-16 data revealed that theB. abortuspopulation segregated into two distinct groups (B and C) (Figure 3B), with African strains clustering in abortus B, while strains from Asia, the Americas, and Europe were predominantly found in abortus C (Figure 3B). Within C I, identical MLVA-16 genotypes were shared among strains from the USA, Costa Rica, Kazakhstan, Italy, and Portugal (Figure 3B). In C II, the majority of shared MLVA-16 genotypes were observed between strains from the USA and Portugal, USA and Brazil, and Bangladesh and Brazil (Figure 3B).
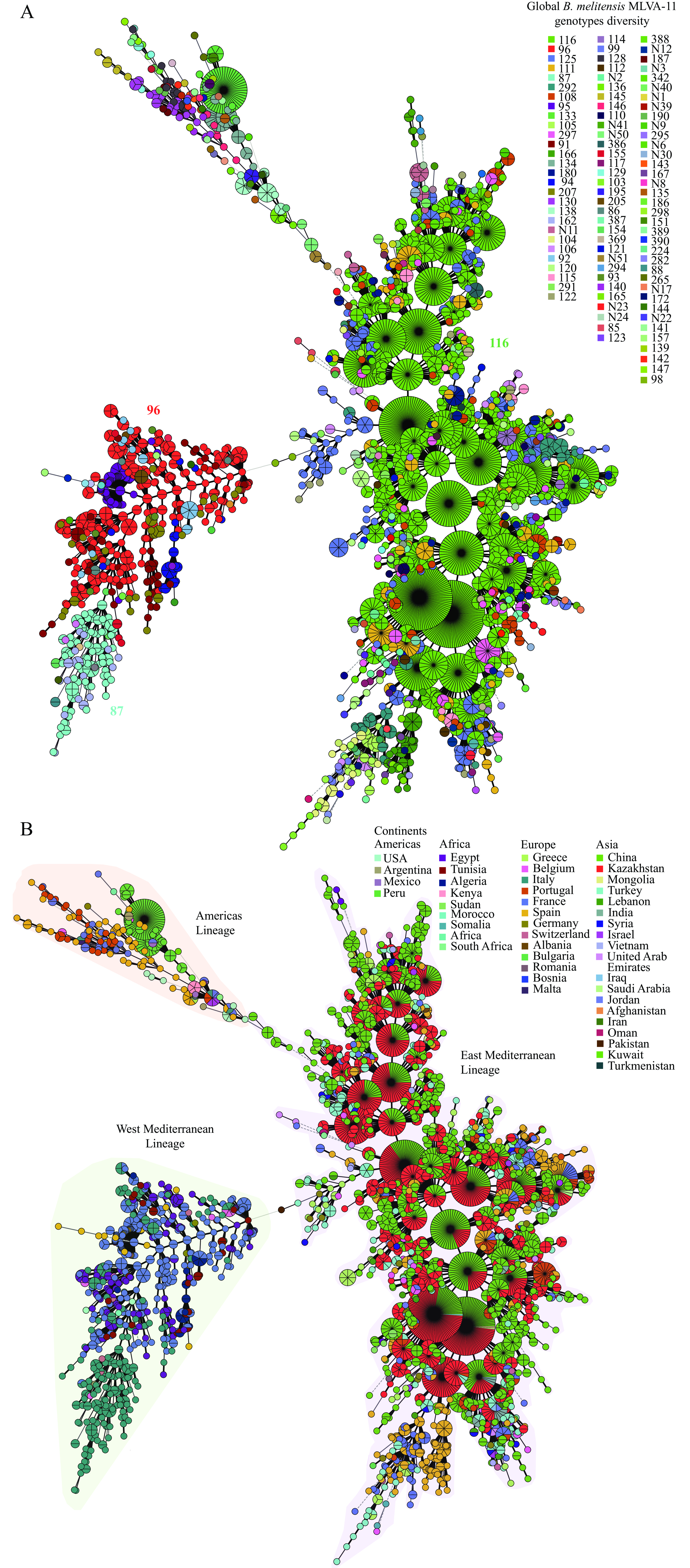
Within theB. melitensis, 216 MLVA-11 genotypes were identified, with five predominant genotypes (GTs): 116, 96, 125, 111, and 87, accounting for 54.4% (2,733/5,019), 7.1% (360/5,019), 6.1% (307/5,019), 3.6% (182/5,019), and 2.2% (112/5,019), respectively (Figure 4A). GT116 was distributed across at least 18 countries, GT96 was identified in nine countries, GT125 was present in 20 countries
.  Figure 4.
Figure 4.MLVA-11 genotypes diversity (A) and MLVA-16 genetic relationships (B) of globalB. melitensisstrains.
Note: (A) Color marks the MLVA-11 genotypes; numbers in the figure indicate the dominant genotypes. (B) Color-coded countries in which strains were isolated; all B. melitensisstrains were divided into three lineages: East Mediterranean, West Mediterranean, and Americas.Minimum spanning tree analysis revealed that theB. melitensisclustered into three distinct lineages: Eastern Mediterranean, Western Mediterranean, and Americas (Figure 4B). The Eastern Mediterranean strains predominated in Asia and Europe; the Western Mediterranean lineage comprised strains from Italy, France, Egypt, and Algeria; The American lineage encompassed strains from the USA, Peru, Spain, and Portugal (Figure 4B). Among the three lineages, the Eastern Mediterranean exhibited the highest frequency of shared MLVA-16 genotypes (Figure 4B).
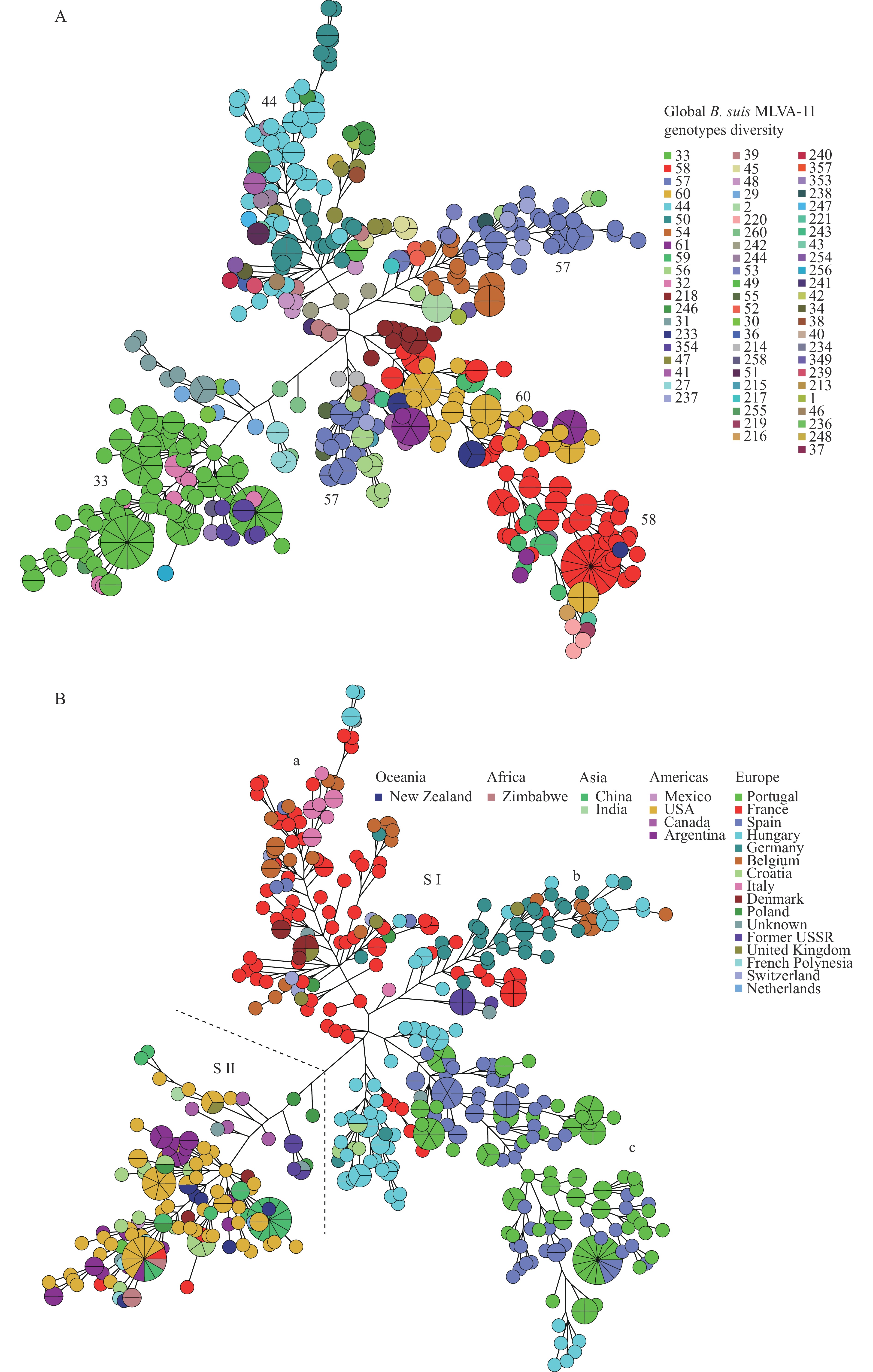
In theB. suispopulation, analysis revealed 67 distinct MLVA-11 genotypes, with five dominant circulating genotypes: GT33 (22.2%, 126/592), GT58 (12.6%, 75/592), GT57 (9.2%, 55/592), GT60 (7.9%, 47/592), and GT44 (7.7%, 46/592) (Figure 5A).
 Figure 5.
Figure 5.MLVA-11 genotype diversity (A) and MLVA-16 genetic relationships (B) of globalB. suisstrains.
Note: (A) Color marks the MLVA-11 genotypes; numbers in the figure indicate the dominant genotypes; (B) Color-coded countries in which strains were isolated; all B. suisstrains were divided into two lineages (S I and S II), and further S I was further sorted into three subgroups (a-c).Minimum spanning tree analysis demonstrated thatB. suisstrains clustered into two major lineages (SI and SII), with SI further subdividing into three distinct sub-clades (a−c) (Figure 5B). Notably, shared MLVA-16 genotypes were observed in SI sub-clade b (Figure 5B), while in SII, a single shared MLVA-16 genotype was identified (Figure 5B).
-
Brucellastrains exhibit widespread distribution across six continents, the extensive spread and dispersal of these pathogens has been facilitated by frequent livestock exchange and trade (4). The paucity of comprehensive surveys and research in Africa presents a significant obstacle to understanding the disease. Consequently, successful prevention, control, and eradication of brucellosis in low-income countries necessitates substantial financial support, unwavering commitment, and sustained long-term programs. The expanding host spectrum of theBrucellaspp. population is a critical factor in its ecological persistence and maintenance. Active spillover between domestic animals and wildlife is increasingly recognized as a potential source of human infection. WhileBrucellaspp. occasionally colonize non-preferred hosts, there remains high potential for discovering additional ecologically significant natural hosts (5).
Global phylogenomic analysis reveals an African origin forB. abortus, with subsequent spread to the Middle East, Europe, and Asia, likely facilitated by infected cattle movement (6).B. abortusstrains from Kazakhstan and Russia show genetic relationships with Portuguese, Brazilian, and US isolates, suggesting ancient lineage dispersal from Europe westward to South America and eastward to Turkey, Russia, and Asia (7).
TheB. melitensispopulation exhibits the highest genetic diversity, with particularly significant genetic homogeneity observed within the E. Mediterranean lineage, especially among Asian strains. All Asian strains clustering into genotype II alongside SEA strains (8). the spread ofB. melitensissubgenotype IIi from Central Asian countries to Russia likely occurred via the northern route of the Great Silk Road, which connected eastern countries with Northern Europe (9). The global trade and movement of ruminants has facilitated the spread and dispersal ofB. melitensis, necessitating stricter regulations on animal transfers from high-epidemic areas and enhanced cross-border inspection and quarantine protocols.
B. suisstrains exhibit significant genetic heterogeneity across different global territories. The maintenance and spread ofB. suisbiovar 2 in Europe represents a dynamic process linked to natural wild boar expansion as the primary wild reservoir, while long-distance transmission largely depends on human activities (10). Surveillance and control measures in endemic European and Asian regions are essential to accurately assess its public health risk.
This study provides novel insights into the global species/biovars, host spectrum, and genetic diversity ofBrucellaspp. However, several limitations warrant consideration. First, our reliance on international MLVA database data may present an incomplete distribution overview, necessitating further investigation. Second, the complex transmission dynamics of brucellosis demand more nuanced analysis of the interplay between human behavior, environmental factors, and microbial genetics in disease transmission.
The global distribution ofBrucellaspecies exhibits remarkable genetic and phenotypic diversity, characterized by extensive host range adaptation and broad territorial spread, presenting significant challenges for worldwide surveillance and control efforts. A critical impediment to effective brucellosis management in low-income countries remains the limited allocation of governmental and regional resources. These findings emphasize the urgent need to establish a comprehensive global pathogen surveillance system and molecular tracking network platform to elucidate the composition of circulatingBrucellastrains and understand the global transmission patterns of brucellosis.
-
All researchers who have previously published data on the isolation, identification, and molecular characterization ofBrucellaspp. worldwide. The developers and administrators of the internationalBrucellaMLVAbank genotyping databases (Brucella v4_6_5).
HTML
| Citation: |

|