
 Download:
Download:




-
Campylobacter jejuni(C. jejuni)
is a prevalent foodborne zoonotic pathogen and has been identified as the primary cause of human gastroenteritis globally (1-2). Besides gastrointestinal illness,C. jejuniis also associated with autoimmune conditions such as Guillain–Barré syndrome and Miller Fisher syndrome (1,3). Although infections caused byC. jejuniare often self-limiting and typically resolve within a few days without antibiotic intervention, effective antimicrobial treatment is essential for immunocompromised patients or severe cases of the disease. The rapid increase in antimicrobial resistance (AMR) amongC. jejunistrains has become a critical public health concern. Consequently, the World Health Organization has listed fluoroquinolone-resistantCampylobacterspp. as one of the six high-priority antimicrobial-resistant pathogens posing the greatest threat to human health (4). Whole genome sequencing (WGS) has the capacity to generate vast amounts of precise data rapidly, which can subsequently be utilized for species identification, typing, phylogenetic analyses, and determining virulence and resistance characteristics (5). WGS is progressively utilized as the foremost approach for foodborne pathogen surveillance in public health laboratories, supplanting prior conventional typing methods.
Recently,Campylobacterspp. has emerged as the most prevalent bacteria causing human diarrhea in Beijing, China, with an alarmingly high burden on human health (6). Only a few reports have analyzed the genetic diversity and AMR profiles of clinical strains ofCampylobacterin Beijing (6–7), and such studies have lacked the resolution provided by WGS-based typing methods. Therefore, larger genomic epidemiology studies are needed to elucidate the molecular characteristics ofCampylobacterstrains circulating in Beijing and more broadly in China.
In this investigation, a total of 184C. jejunistrains were collected from patients with diarrhea during active surveillance in Beijing throughout a three-year period (2019–2021). This study presents a comprehensive genomic analysis, examining the genetic diversity, virulence potential, and AMR profiles of the isolated strains.
-
Hospital-based active surveillance ofCampylobacterhas been conducted since 2010 in Beijing, China. ACampylobacterisolation kit incorporating a membrane filter method (ZC-CAMPY-002, Sinova Biotechnology Co., Ltd., Qingdao, China) was employed to isolateCampylobacter. In brief, a 1 mL stool specimen suspension was transferred into 4 mL of enrichment medium provided in the kit. The enriched suspension was incubated at 37 ℃ for 24 hours under microaerophilic conditions, consisting of 5% O2, 10% CO2, and 85% N2. Approximately 300 µL of the enriched culture was spotted onto the membrane filter surface and then pasted onto both Karmali and Columbia agar plates. The plates were subsequently incubated in a microaerophilic atmosphere at 37 ℃ for 48 hours. Suspected colonies were picked and identified using a real-time PCR assay targeting thehipOgene ofC. jejuni. From 2019 to 2021, 184C. jejuniisolates were obtained from this surveillance program.
-
The minimum inhibitory concentration (MIC) of allC. jejuniisolates was determined using the agar dilution method recommended by the Clinical and Laboratory Standards Institute document (CLSI M45-P). Six classes of 11 antimicrobial agents (Sinova Biotechnology Co., Ltd., Qingdao, China) were used for AST: erythromycin (ERY), azithromycin (AZI), nalidixic acid (NAL), ciprofloxacin (CIP), gentamicin (GEN), streptomycin (STR), chloramphenicol (CHL), florfenicol (FLO), tetracycline (TET), telithromycin (TEL), and clindamycin (CLI). The breakpoints for resistance used in this study were based on standards used in the National Antimicrobial Resistance Monitoring System (NARMS), except for ERY, CIP, and TET, which were based on CLSI guidelines. The following MIC values were determined forC. jejuni: ERY ≥32 μg/mL, AZI ≥1 μg/mL, NAL ≥32 μg/mL, CIP ≥4 μg/mL, GEN ≥4 μg/mL, STR ≥16 μg/mL, CHL ≥32 μg/mL, FLO ≥8 μg/mL, TET ≥16 μg/mL, TEL ≥8 μg/mL, and CLI ≥1 μg/mL.C. jejuniATCC 33,560 was included in the test as a quality control strain. Multidrug resistance was defined as resistance to at least three classes of antimicrobials in this study.
-
DNA was extracted utilizing a QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). The quantification of the extracted genomic DNA (gDNA) was determined through agarose gel electrophoresis and fluorometric analysis (Qubit 2.0). WGS was performed on an Illumina PE150 platform with 100× coverage (Novogene Technology Co., Ltd., Beijing, China). Raw sequencing data were assessed for quality, trimmed, and subsequently assembledde novointo a draft genome sequence using the SPAdes 3.13 software.
Multilocus sequence typing (MLST) was conducted utilizing WGS data in accordance with theCampylobacterPubMLST scheme (
https://pubmlst.org/ ). STs and CCs were identified for each isolate. AMR genes were analyzed using the NCBI AMRFinderPlus tool 3.1.1b (https://ftp.ncbi.nlm.nih.gov/pathogen/Antimicrobial_resistance/AMRFinder/ ). Virulence genes were detected via the virulent factors of pathogenic bacteria (VFDB) database (http://www.mgc.ac.cn/cgi-bin/VFs/v5/main.cgi ). Whole genome single nucleotide polymorphisms (wgSNPs) analysis was performed on all draft genomes with the reference strain NCTC 11168 (GenBank ID: NC_002163.1) using the parsnp software. Finally, the phylogenetic tree and heatmap of resistance and virulence genes were visualized using the ChiPlot tool (https://www.chiplot.online/ ). -
MLST analysis of the 184C. jejuniisolates produced 71 distinct STs, categorized into 19 CCs and unassigned (not belonging to any known CC) categories (
Supplementary Table S1 ). Out of these, 23 isolates belonged to 16 unassigned STs. The most prevalent STs were ST760 (6.5%), followed by ST49 (6.0%) and ST22 (5.4%). Thirty STs were represented by only one isolate each. The most common CC, CC21 (24.5%), consisted of 11 STs: ST21, ST50, ST298, ST760, ST1811, ST3597, ST6500, ST8261, ST9873, ST9960, and ST10075. The second most frequent CC, CC45, comprised six STs and 13 isolates (7.1%). The remaining CCs included relatively few STs or a small number of strains. -
In the present study, 13C. jejuniisolates (7.1%) demonstrated susceptibility to all 11 tested antimicrobials. The highest recorded resistance rates were observed for CIP (76.6%), with NAL (76.1%) and TET (71.2%) following closely behind. Conversely, resistance rates for ERY, TEL, and CHL were relatively low at 3.8%, 7.6%, and 8.2% respectively. The resistance rates for other antimicrobials included FLO (27.3%), CLI (18.5%), STR (11.4%), and both AZI and GEN (10.9%) (Table 1).
Class Antibiotic agent MIC (μg/mL) Resistant strains AMR gene Breakpoint Range ≤0.25 0.5 1 2 4 8 16 32 64 >64 n(%) Macrolides ERY ≥32 0.5–64 84 69 14 7 1 2 1 2 4 7 (3.8) 50S rRNA_L22_A103V 0 23S rRNA_A2075G 1 1 cmeABC 2 3 5 AZI ≥1 0.5–64 164 5 2 6 1 1 1 2 2 20 (10.9) 50S rRNA_L22_A103V 1 1 23S rRNA_A2075G 1 1 cmeABC 3 2 4 1 1 1 1 2 15 Quinolones NAL ≥32 0.5–64 3 19 6 4 8 4 5 36 99 140 (76.1) gyrA_T86I 5 34 98 137 cmeABC 3 21 58 82 CIP ≥4 0.5–64 17 15 11 5 26 34 41 22 13 141 (76.6) gyrA_T86I 5 26 34 39 22 12 138 cmeABC 3 25 27 11 11 11 88 Aminoglycosides GEN ≥4 0.5–64 144 13 7 4 0 3 2 3 8 20 (10.9) aph(3')–IIIa 2 1 1 2 6 aph(2'')–If 1 1 3 5 aac(6')–Ie/aph(2'')–Ia 2 1 3 STR ≥16 0.5–64 104 23 23 4 9 6 4 5 6 21 (11.4) ant(6)-Ia 1 1 2 aadE 1 1 Tetracyclines TET ≥16 0.5–64 28 12 6 3 4 4 7 37 83 131 (71.2) tetO 3 7 35 78 123 cmeABC 4 5 23 46 78 Phenicols CHL ≥32 0.5–64 16 27 25 43 21 37 12 1 2 15 (8.2) FLO ≥8 0.5–64 25 24 50 35 30 8 8 4 50 (27.3) Ketolides TEL ≥8 0.25–32 45 33 30 50 12 4 3 3 4 14 (7.6) Lincosamides CLI ≥1 0.25–32 107 43 16 4 4 1 4 1 4 34 (18.5) Abbreviation: MIC=minimum inhibitory concentration; AMR=antimicrobial resistance;C. jejuni=Campylobacterjejuni; ERY=erythromycin; AZI=azithromycin; NAL=nalidixic acid; CIP=ciprofloxacin; GEN=gentamicin; STR=streptomycin; TET=tetracycline; CHL=chloramphenicol; FLO=florfenicol; TEL=telithromycin; CLI=clindamycin. * The gray shade indicates resistance strains and red numbers indicate the number of resistance strains. Table 1.MIC distribution and AMR phenotype and genotype in 184C. jejunistrains.
Regarding multidrug resistance, 77C. jejuniisolates (41.8%) were resistant to three or more classes of antimicrobials and showed 43 different resistance patterns. Among these, the predominant resistance pattern was NAL-CIP-TET-FLO (12.0%). Three isolates were resistant to at least 10 antimicrobials (
Supplementary Figure S1 ). -
Genomic analysis revealed a total of 29 AMR determinants among 184C. jejuniisolates. These included 26 acquired AMR genes and three resistance-conferring point mutations (Table 2). ThegyrA_T86I point mutation, associated with resistance to quinolones, was the most prevalent, identified in 96.2% of the isolates. ThetetOgene, conferring resistance to tetracycline, was also highly prevalent, found in 82.6% of isolates.
Genes Class Gene No. of isolates (%) Resistance genes Quinolones gyrA_T86I 177 (96.2) Tetracyclines tetO 152 (82.6) ß-Lactams Any of the following 147 (79.9) blaOXA-193 74 (40.2) blaOXA-461 19 (10.3) blaOXA-591 17 (9.2) blaOXA-184 12 (6.5) blaOXA-460 9 (4.9) blaOXA-592 7 (3.8) blaOXA-583 3 (1.6) blaOXA-631 3 (1.6) blaOXA-465 1 (0.5) blaOXA-594 1 (0.5) blaEC 1 (0.5) Macrolides Any of the following 37 (20.1) 50S rRNA_L22_A103V 35 (19.0) 23S rRNA_A2075G 2 (1.1) Aminoglycosides Any of the following 67 (36.4) aph(3')-IIIa 18 (9.8) ant(6)-Ia 14 (7.6) sat4 12 (6.5) aph(2'')-If 9 (4.9) aac(6')-Ie/aph(2'')-Ia 5 (2.7) aadE 4 (2.2) aad9 1 (0.5) Phenicols catA13 2 (1.1) Arsenics arsP 95 (51.6) acr3 73 (39.7) Lincosamides InuC 6 (3.3) Multidrug efflux pump cmeA 183 (99.5) cmeB 114 (62.0) cmeC 184 (100.0) cmeABC 114 (62.0) Virulence genes Invasion ciaB; ciaC; flhA; flhB; fliP; fliQ; fliR; 184 (100.0) flaC 182 (98.9) Adhesion cadF; jlpA; pebA; 184 (100.0) porA 112 (60.9) Toxin cdtB 184 (100.0) cdtA 182 (98.9) cdtC 182 (98.9) Chemotaxis cheA; cheV; cheW; cheY; 184 (100.0) Motility flgG; flgH; fliF; fliM; 184 (100.0) fliY 183 (99.5) flgI 183 (99.5) flgE 181 (98.4) fliK 178 (96.7) flaB 22 (12.0) flaA 21 (11.4) LOS hldD; waaC; 184 (100.0) htrB 183 (99.5) gmhA 66 (35.9) hldE 55 (29.9) cstIII 41 (22.3) neuA 41 (22.3) neuB 41 (22.3) neuC 41 (22.3) CPS kpsS 183 (99.5) kpsD 183 (99.5) kpsE 177 (96.2) Cj1417c 158 (85.9) Cj1419c 158 (85.9) Cj1420c 154 (83.7) waaF 130 (70.7) kpsF 71 (38.6) kpsT 36 (19.6) Abbreviation: AMR=antimicrobial resistance;C. jejuni=Campylobacter jejuni; LOS=lipo-oligosaccharide; CPS=capsule polysaccharide. Table 2.Distribution of AMR and virulence genes in 184C. jejunistrains.
A variety of 11 knownblaOXAvariants were detected in 147 strains (79.9%), withblaOXA-193,blaOXA-461, andblaOXA-591being the most prevalent (40.2%, 10.3%, and 9.2%, respectively). In contrast, a much smaller fraction of strains harbored resistance markers for other antimicrobials. Resistance to macrolides via two point mutations,50S rRNA_L22_A103V and23S rRNA_A2075G, was observed in 19.0% and 1.1% of isolates, respectively.
Additionally, seven genes for resistance to aminoglycosides (i.e.,aph(3')-IIIa,ant(6)-Ia,sat4,aph(2'')-If,aac(6')-Ie/aph(2'')-Ia,aadE, andaad9) were detected in fewer than 10% of the isolates. High prevalence of arsenic resistance genes,arsPandacr3, was observed. Notably, thecmeABCoperon, encoding a multidrug efflux pump and consisting ofcmeA,cmeB, andcmeCgenes, was present in 62% ofC. jejuniisolates. The prevalence of remaining resistance genes was no greater than 3.3%. Detailed prevalence rates of the aforementioned AMR genes can be found inTable 2.
-
Of the seven strains exhibiting ERY-resistant phenotypes, one strain carried theA2075Gmutation in the23S rRNAgene, and five strains contained thecmeABCmulti-efflux pump operon gene (Table 1). Among the 20 strains with AZI-resistant phenotypes, one displayed the23S rRNA_A2075Gmutation, another exhibited the50S rRNA_L22_A103V mutation, and fifteen possessed thecmeABCoperon. These two isolates with the23S rRNA_A2075Gpoint mutation demonstrated high-level resistance to macrolides (ERY, AZI MIC >64 μg/mL).
Out of 141 CIP-resistant strains, 97.9% (n=138) presented thegyrA_T86I mutation, and 62.4% (n=88) carried thecmeABCoperon. Interestingly, thegyrA_T86I point mutation was present across the entire range of CIP resistance (MIC 4–64 μg/mL). Of the 131 TET-resistant strains, 93.9% (n=123) harbored thetetOgene. Moreover, 59.5% (n=78) of the TET-resistant strains contained thecmeABCoperon.
-
Virulome analysis identified 47 virulence genes across 184C. jejuniisolates. These genes were classified into seven categories based on their roles in pathogenesis/colonization (Table 2). The prevalence rates of cell invasion-associated genes included:ciaB,ciaC,flhA,flhB,flip,fliQ, andfliR(all at 100%), andflaC(98.9%). Adherence and colonization-related genes demonstrated prevalence rates ofcadF,jlpA,pebA(all at 100%), andporA(60.9%). The cytolethal distending toxin (CDT) genes encoded by thecdtABCoperon showed prevalence rates ofcdtB(100%) andcdtAandcdtC(both at 98.9%). All 184 isolates contained chemotaxis-associated genes:cheA,cheV,cheW, andcheY. Genes involved in motility displayed prevalence rates of:flgG,flgH,fliF,fliM(all at 100%),flgI,fliY(both at 99.5%),flgE(98.4%),fliK(96.7%),flaB(12.0%), andflaA(11.4%). Moreover, the analysis identified various genes implicated in the biosynthesis of lipo-oligosaccharide (LOS) and capsule polysaccharide (CPS). Percentages of all detected virulence genes categorized by function are outlined inTable 2.
-
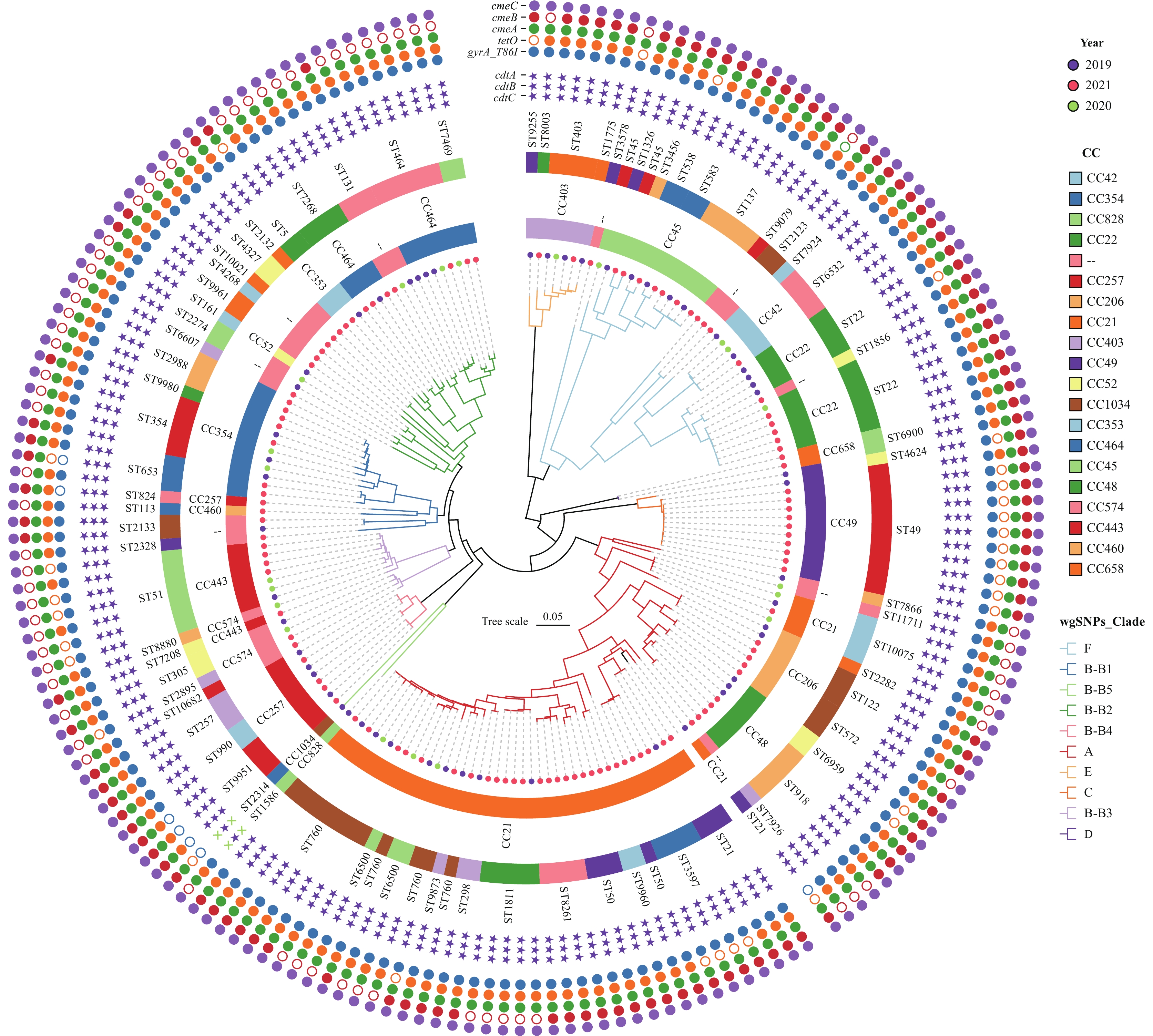
The wgSNP phylogenetic analysis of 184C. jejunistrains identified six major clades, designated A–F (Figure 1), with no association between the lineages and the years of isolation. Clade A consisted of 62 strains from CC21, CC206, or CC48, while the largest clade, clade B, included 68 isolates forming five sub-clusters (B1–B5) with diverse CCs as follows: B1: CC257, CC354, CC460; B2: CC52, CC353, CC464; B3: CC443, CC574; B4: CC257; B5: CC828, CC1034. Notably, CC257 appeared in both B1 and B4, indicating the differentiation and expansion of clonal groups. Lineages C, D, and E each contained unique independent CCs, specifically CC49, CC58, and CC403, respectively. Additionally, 33 strains from CC22, CC42, or CC45 formed a single lineage, lineage F.
 Figure 1.
Figure 1.Phylogenetic analysis of 184C. jejunistrains in Beijing from 2019 to 2021.
Note: The phylogenetic tree is based on wgSNPs analysis. CCs and STs are shown in colored rings for each strain, and tree branches are color coded to highlightC. jejunistrains from clades A, C, D, E, F, and sub-cluster of clade B (B1-B5). The isolation year of each strain is indicated by colored dots. Unassigned CC is denoted by two short lines (--). The presence of 5 main resistance genes is denoted by colored solid circles:gyrA_T86I (blue),tetO(orange),cmeA(green),cmeB(red) andcmeC(purple). The absence is indicated by hollow circles. The presence of toxin genes (cdtA,cdtBandcdtC) is denoted by purple star symbol and the absence is denoted by cross symbol.
Abbreviation:C. jejuni=Campylobacterjejuni; wgSNPs=whole genome single nucleotide polymorphisms; CC=clonal complex; ST=sequence type.
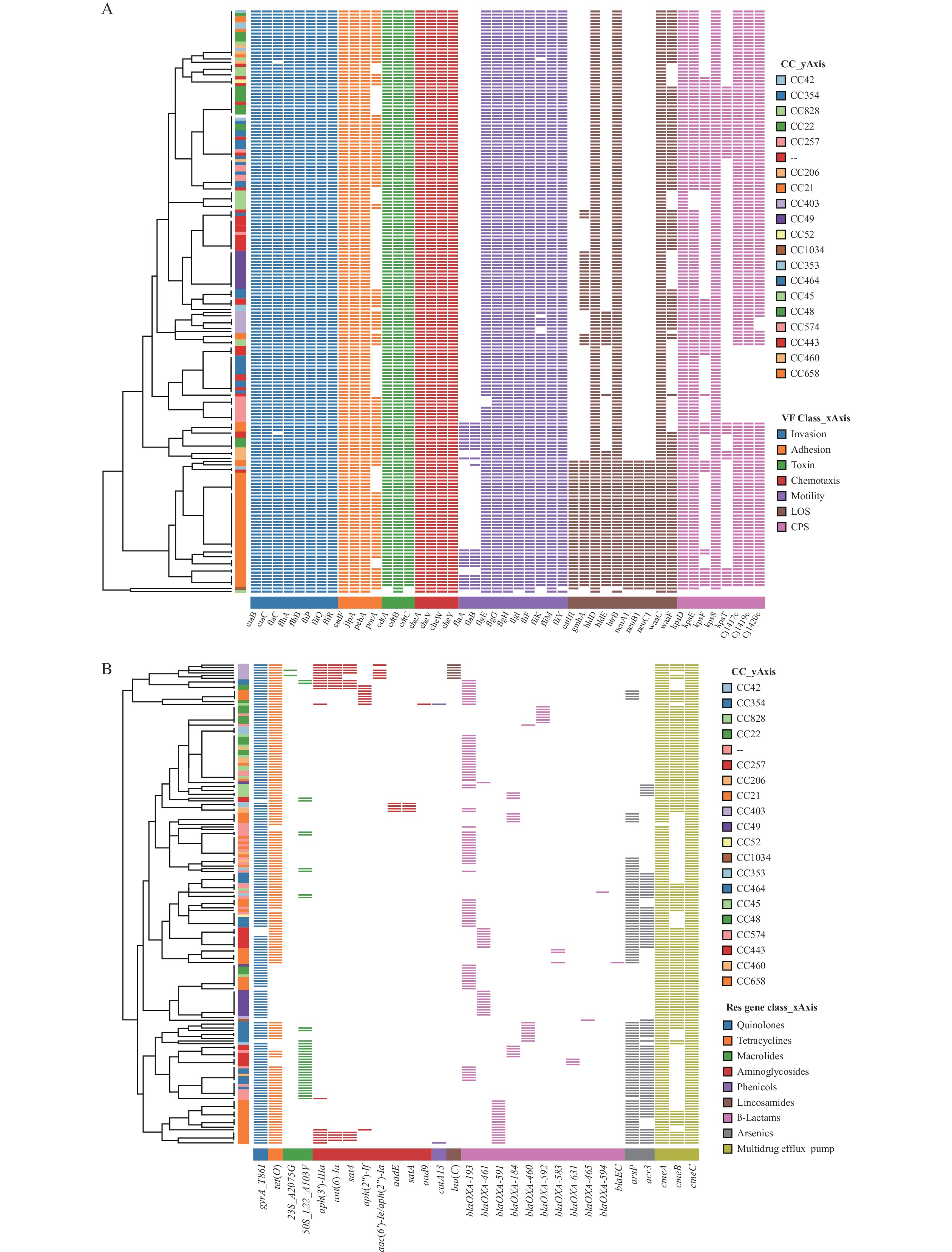
The heatmap of virulence genes revealed that all strains were divided into two clusters related to CC, based on the distribution of virulence factors. Strains from CC21 exhibited a wide distribution of virulence determinants, particularly those related to LOS (Figure 2A). CC354 and CC257 strains possessed fewer CPS genes compared to other strains. Although no correlation was found between the observed AMR genes and their distribution of CCs on the heatmap (Figure 2B), interestingly, 12 isolates from CC49 displayed the least resistance genes and were sensitive to most antimicrobial agents.
 Figure 2.
Figure 2.Annotation heatmap of virulence and AMR genes among 184C. jejunistrains in Beijing from 2019 to 2021. (A) Virulence genes; (B) AMR genes.
Note: The color strips of Y-axis represent CCs corresponding to each strain. The color blocks of X-axis represent the categories of genes. Binary heatmaps show the presence and absence of virulence and AMR genes. Colored cells represent the presence of genes and white cells represent the absence of the genes. Unassigned CC is denoted by two short lines (--).
Abbreviation: AMR=antimicrobial resistance;C. jejuni=Campylobacterjejuni; CC=clonal complex; VF=virulence factors.
-
In recent decades, the dramatic increase in AMR inCampylobacterworldwide has prompted research on the prevalence and molecular determinants of resistance. In this study,C. jejuniisolates exhibited a high rate of resistance to certain antimicrobials (CIP, NAL, and TET) and a low rate of resistance to ERY, which concurs with findings previously reported in China (8) and elsewhere (9–12). The high percentage of quinolone-resistantC. jejunistrains (97.9%) with point mutationgyrA_T86I was frequently observed, which contributed to the high-level resistance to quinolones (9–10,12). ThetetOgene was detected in 93.9% of TET-resistantC. jejuniisolates, which also corroborates previous studies (9–10,12). The two markers of AMR to macrolides,50S rRNA_L22_A103V and23S rRNA_A2075G, were detected at low prevalence (19.0% and 1.1%, respectively) amongCampylobacterisolates. Notably,C. jejuniisolates (62.0%) harbored thecmeABCoperon, which encodes a member of the resistance-nodulation cell division superfamily of multidrug efflux transporters and functions synergistically with other mechanisms to contribute to resistance to quinolones, macrolides, and tetracyclines (9–10). In this study, three strains with phenotypic resistance to quinolones did not present the mutation ingyrA_T86I; however, the presence of thecmeABCoperon was observed, indicating that the efflux pumps could be conferring resistance to quinolones in these three strains. Also, six ERY-resistant strains did not carry the mutationA2075Gin the23S rRNAgene; however, the presence of thecmeABCoperon was observed in five strains, which would likely be responsible for the resistance to ERY in strains that did not present the mutation in the23S rRNA. Finally, eight TET-resistant strains did not harbor thetetOgene; similarly, the presence of thecmeABCoperon was observed in all eight strains, which would also be responsible for the resistance to TET. Taken together, these data contribute to our understanding of theCampylobacterresistome, which will support the development of AMR surveillance programs in China.
The mechanisms underlying the pathogenic processes by whichCampylobacterspecies cause diarrhea remain unclear (1). This study found that a large proportion ofCampylobacterstrains possessed genes linked to bacterial motility, invasion, adhesion, and chemotaxis to epithelial cells, all of which play vital roles in the onset ofCampylobacterinfection (2). These results support earlier research indicating that adherence, colonization, and invasion genes (e.g.,cadF,ciaB,ciaC,flaC,jlpA,pebA,porA) are highly conserved amongC. jejunistrains and present in the majority of clinical isolates (13–14), emphasizing the potential virulence of theseCampylobacterstrains in causing human infections.
Furthermore, virulence marker determinantscdtABC, which encode CDT and significantly contribute to diarrhea by interfering with the division and differentiation of intestinal crypt cells, were also detected in most examined isolates (>98.9%). Previous investigations have reported a high prevalence of CDT inCampylobacterstrains isolated from patients experiencing life-threatening diarrhea (10,14–15). While the functions of individual LOS genes (cstIII,gmhA,hldD,hldE,trB,neuA,neuB,neuC,waaC, andwaaF) have yet to be clearly established, prior studies have suggested that these genes are crucial for forming human ganglioside-like LOS structures capable of inducing Guillain–Barré syndrome (3). In this study, the majority of strains carried at least four genes associated with LOS biosynthesis.
This study emphasized the extensive diversity of clinicalC. jejuniisolates circulating in Beijing. Our findings demonstrated that 184C. jejunistrains were classified into 71 STs, with strains belonging to CC21 as the predominant group. CC21 is the largest and most widely distributed CC globally, representing 17.9% of allC. jejunistrains submitted to the PubMLST database. Numerous studies have reported varying major CCs ofCampylobacterin different countries and regions, but CC21, CC45, CC48, and CC353 are consistently the predominant CCs among isolates in many investigations. The high prevalence of ST760 in our study was unexpected, given its scarce representation within the PubMLST database (0.033%, as of February 2023). ST760 was first reported in 2000 from a human gastroenteritis case in England. Subsequently, it spread to several neighboring European countries but not to North or South America. In Asia, there were four reports of ST760 in Jiangsu Province in China in 2006, but no reports in other regions of China or other Asian countries (based on the PubMLST database). These data highlight the wide-range transmission of these strains across regions and indicate that ST diversity varies among countries and regions.
Previous research has demonstrated that associations between clades and the presence of resistance and virulence determinants are prevalent, although not all clades within a phylogenetic tree are characterized by factors such as geographic distribution or year of isolation (11). To date, few studies have focused on the phylogenetic analysis ofCampylobacterstrains in Beijing. In this study, wgSNPs phylogenetic analysis revealed that theC. jejunipopulation from diarrheal patients in Beijing encompasses a wide range of lineages and genotypes, with strains belonging to the same ST and/or CC grouping together in distinct clusters. Although this study presents limited literature regarding virulence factors linked to specific CCs or STs, it emphasizes the virulence potential of the investigatedCampylobacterisolates. Additionally, one limitation of this study is that allCampylobacterstrains were isolated from human samples, with none originating from alternative sources such as food or animals.
In conclusion, this study represents one of the most extensive genomic analyses ofC. jejuniin Beijing, offering valuable insights into the prevalence of virulence genes, AMR markers, as well as the phylogenetic relationships and circulating genotypes from 2019 to 2021. Moreover, we report the high resistance rates to quinolone and tetracycline alongside the low resistance rate to erythromycin among theC. jejunistrains identified in Beijing. This information proves crucial for the development of monitoring, control, and prevention strategies to address the growing concern of resistance posed by this pathogen.
-
No conflicts of interest.
CC type ST type No. of isolates (%) CC type ST type No. of isolates (%) CC type ST type No. of isolates (%) CC21 ST21 4 (2.2) CC52 ST161 1 (0.5) CC574 ST305 2 (1.1) ST50 4 (2.2) CC206 ST122 3 (1.6) ST2895 1 (0.5) ST298 2 (1.1) ST572 3 (1.6) ST8880 1 (0.5) ST760 12 (6.5) ST2282 1 (0.5) ST10682 1 (0.5) ST1811 5 (2.7) CC257 ST257 3 (1.6) CC658 ST6900 2 (1.1) ST3597 4 (2.2) ST824 1 (0.5) CC828 ST1586 1 (0.5) ST6500 3 (1.6) ST990 2 (1.1) CC1034 ST2314 1 (0.5) ST8261 4 (2.2) ST9951 3 (1.6) unassigned ST131 3 (1.6) ST9873 1 (0.5) CC353 ST5 2 (1.1) ST1856 1 (0.5) ST9960 2 (1.1) ST2132 1 (0.5) ST2123 2 (1.1) ST10075 4 (2.2) CC354 ST354 5 (2.7) ST2133 2 (1.1) CC22 ST22 10 (5.4) ST653 3 (1.6) ST2274 2 (1.1) CC42 ST6532 4 (2.2) ST2988 3 (1.6) ST2328 1 (0.5) ST7924 1 (0.5) ST9980 1 (0.5) ST3578 1 (0.5) CC45 ST45 2 (1.1) CC403 ST403 4 (2.2) ST4268 1 (0.5) ST137 5 (2.7) ST1775 1 (0.5) ST4327 2 (1.1) ST538 2 (1.1) ST8003 1 (0.5) ST6607 1 (0.5) ST583 2 (1.1) ST9255 1 (0.5) ST7866 1 (0.5) ST1326 1 (0.5) CC443 ST51 7 (3.8) ST7926 1 (0.5) ST3456 1 (0.5) ST7208 1 (0.5) ST9079 1 (0.5) CC48 ST918 5 (2.7) CC460 ST113 1 (0.5) ST9961 2 (1.1) ST6959 2 (1.1) CC464 ST464 6 (3.3) ST10021 1 (0.5) CC49 ST49 11 (6.0) ST7268 4 (2.2) ST11711 1 (0.5) ST4624 1 (0.5) ST7469 2 (1.1) Abbreviation: CC=clonal complex; ST=sequence type;C. jejuni=Campylobacterjejuni. Table S1.Distribution of CCs and STs in 184C. jejunistrains.
HTML
Sample Collection andCampylobacterIsolation
Antimicrobial Susceptibility Testing (AST)
WGS and Genomic Analysis
Diversity of MLST Genotype STs and CCs
AST
Detection of Antimicrobial Resistance Genes
Correlation of Phenotypic and Genotypic Resistance
Detection of Virulence Factors
Phylogenetic Analysis and Heatmap of AMR and Virulence Genes
| Citation: |

|
