
 Download:
Download:





-
Prion disease (PrD) belongs to neurodegenerative diseases showing many similarities to Alzheimer's disease (AD) and Parkinson's disease (PD). Long incubation time and short clinical duration of PrDs highlight a slow progression at the early stage and rapid deterioration at the late stage of various abnormalities in central nerve system (CNS). In this report, we briefly describe some of the major findings of changes in intracellular components and biological pathways in the brain during prion infection including microtubule-associated proteins (MAPs), microtubule affinity-regulating kinase 4, p21-activated kinases, CK2, Polo-like kinases, etc. as well as hypothesizes some physiological and pathological approaches mainly based on our studies.
-
The similarities in global brain differentially-expressed mRNAs (messenger RNA) and in the biological pathways have been found in different types of human PrDs, such as sporadic Creutzfeldt-Jacob disease (sCJD), fatal familial insomnia with D178N mutation (D178N FFI) and genetic CJD with G114V mutation (G114V gCJD) (1-3). The commonly changed pathways involved signal transduction, ion transport, oxidative phosphorylation, regulation of actin cytoskeleton, MAPK signaling pathway, etc.
Global brain protein profiles in sCJD, FFI, and G114V cases revealed comprehensive disturbances at the terminal stage (4). Many dysregulated biological pathways were commonly identified, such as oxidative phosphorylation, protein export, drug metabolism cytochrome P450, PPAR signaling pathway, and fatty acid metabolism. Analysis of middle and terminal stages of two scrapie agents (139A and ME7) infected mouse models verified more differentially expressed proteins and involved pathways at the final stage. Great similarity in differentially expressed proteins, affected pathways, and biological functions were observed between scrapie-infected mice and human sCJD (5).
Proteomic assays of globalS-nitrosylation (S-nitrosoproteome) in brain tissues of the patients with sCJD, FFI, and G114V gCJD (6) revealed that sCJD contained much more differentially expressed SNO-proteins (DESPs), while FFI had less numbers of DESPs. The most affected pathways were PD, AD, Huntington’s disease, arginine and proline metabolism, and systemic lupus erythematosus. Further study found that the brain levels of nitric oxide (NO) and nitric oxide synthase (NOS) activities of scrapie rodent models started to increase at early stage, reached the peak in the middle stage, and dropped down at late stage (7), which were closely associated with the similar alternative tendencies of many brain proteins.
Proteomic mass spectrometry of global profiles of brain acetylated proteins of scrapie-infected mice showed that overwhelming majority of the differentially expressed acetylated proteins (DEAPs) in mid-early stage was down-regulated, while more portions of DEAPs in mid-late and late stages were up-regulated. Approximately 22.1% of acetylated peptides were mitochondrial-associated. Carbon metabolism, metabolic pathways, biosynthesis of amino acids, glycolysis/gluconeogenesis, pyruvate metabolism, and tricarboxylic acid (TCA cycle) were commonly affected pathways. Overall, 2 major elements in the Sirtuin (Sirt) family, Sirt 1 and Srit 3 were markedly decreased (8-9), which subsequently caused increases of acetylated forms of many down-stream target proteins, such as p53, PGC-1 and STAT3, as well as SOD and ATP5β, and influenced cell viability, scrapie-like prion protein (PrPSc) deposit, cellular ROS level and mitochondria function remarkably.
-
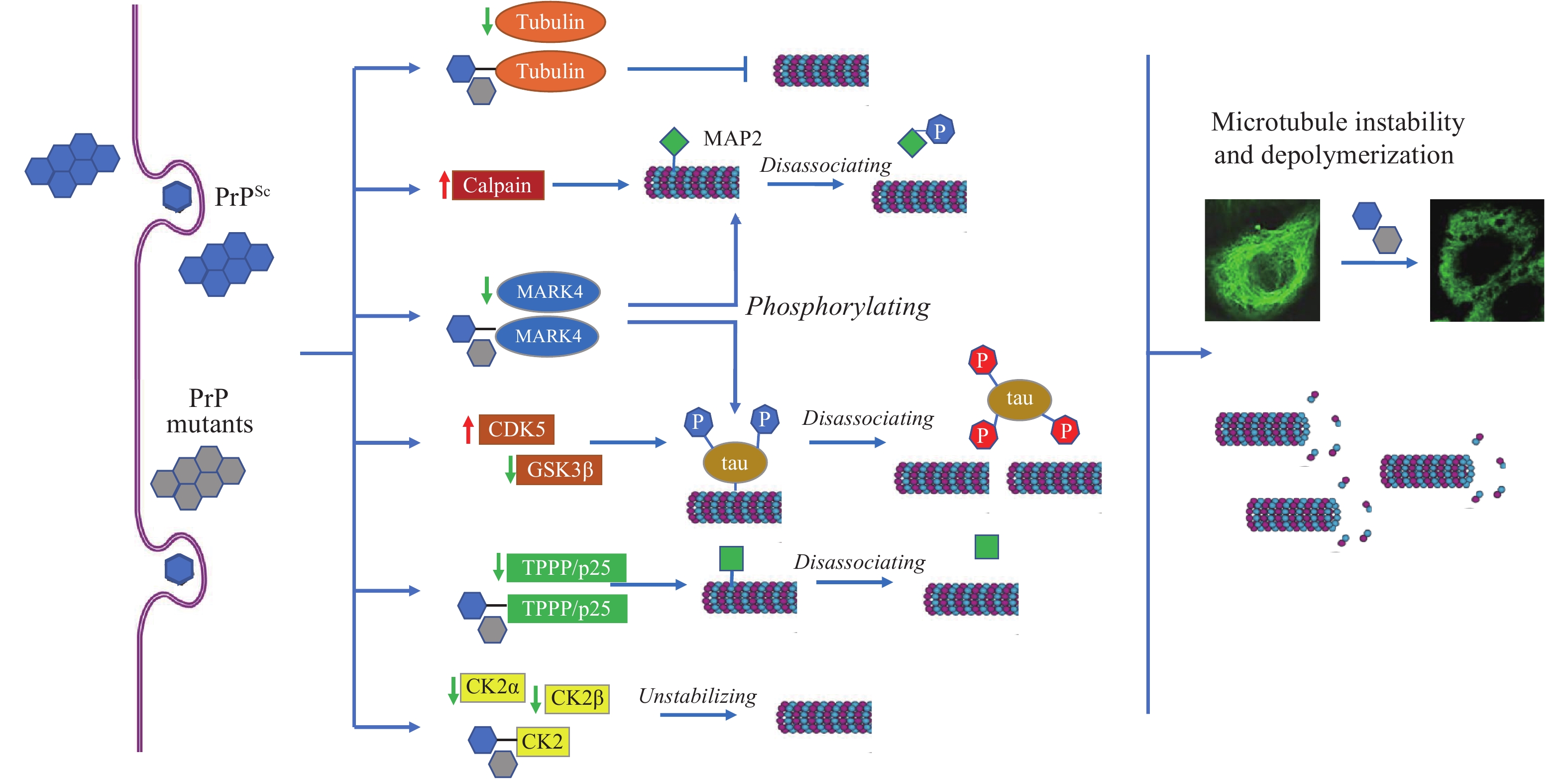
Our serial studies proposed that microtubule disturbance was a common phenomenon in prion infection (Figure 1). Inin vitroassays, prion protein (PrP) revealed actively molecular interaction with tubulin, with the main interacting region within PrP in the domain of five octapeptide repeats (ORs). The wild-type PrP could efficiently stimulate microtubule assemblyin vitroand antagonize Cu2+-induced microtubule-disrupting activityin vivo(10). The gCJD-related PrP mutants with inserted or deleted ORs showed much stronger inhibitive capacities on the microtubule dynamics (11). Expression of Cyto-PrP, a truncated form of PrP removal of its signal peptide and GPI anchor in cultured cells induced remarkable disruption of microtubular structures (12). Knockdown of the expression of PrP via RNA interference antagonized the cytotoxicity of gCJD-associated PrP mutants in cultured cells (13).
 Figure 1.
Figure 1.Schema of prion-induced microtubule instability and depolymerization in brains.
Note: PrPScand PrP mutants accumulate in neurons and induce the abnormal changes in tubulin, calpain, and many MAPs, leading to microtubule instability and depolymerization.
Abbreviation: MAPs=microtubule associated proteins; PrPSc=scrapie-like prion protein; PrP=prion protein, TPPP=Tubulin polymerization promoting protein.
PrP regulates microtubule dynamics via interacting with microtubule-associated proteins (MAPs). Tubulin polymerization promoting protein/p25 (TPPP/p25) is a brain-specific protein stabilizing cellular microtubular ultrastructure. The brain levels of TPPP of scrapie-infected hamsters were significantly reduced. Distinct molecular interactions between TPPP and PrP were identified. Expression of TPPP in cultured cells significantly antagonized the disruption of microtubule structures and rescued the cytotoxicity caused by the accumulation of Cyto-PrP (14).
Tau proteins are also important MAPs in the CNS, showing molecular binding capacity with PrP (15). Tau can interact with tubulin and facilitate microtubule formation.In vitro, PrP showed a notable inhibitive effect not only on the interaction of tau with tubulin, but also on the tau-mediated microtubule formation (16). GSS-related mutant P102L and gCJD-related mutants with 2 and 7 extra ORs showed more active binding capacity with tau than wild-type PrP (17). Inhibition of PrP on tau-enhanced formation of microtubule undergoes, at least partially, through blocking the interaction between tau and microtubule.
Microtubule affinity-regulating kinase 4 (MARK4) is responsible for phosphorylating neuronal MAPs. Significant decreases of brain MARK4 were found in human CJD and scrapie-infected experimental animals, which showed a close correlation with the amounts of PrPSc. Treatment of toxic PrP106-126 peptide onto cultured cells induced a similar reduction of MARK4 and reduction of p-tau at Ser262, indicating that a reduction of MARK4 will result in abnormalities of tau phosphorylation and possibly induce further detachment of microtubules and hinder microtubule transportation (18). We have also identified that MAP2 was markedly decreased in the brains of human prion diseases and scrapie infected rodents. Exposure of the cultured cells to PrP106-126 peptide resulted in a significant reduction of cellular MAP2 and disrupted the microtubule structure. Meanwhile, the level of calpain, which mediates the degradation of a range of cytoskeletal proteins, was significantly increased during prion infection (19).
Protein kinase CK2 is generally composed of 2 catalytic subunits (CK2α or CK2α’) and 2 regulatory subunits (CK2β) and forms a tetrameric structure. The brain levels of CK2α and CK2β decreased in the scrapie-infected experimental rodent models and human PrDs at the terminal stage (20). Strong molecular interaction was observed between PrP and CK2α subunit (21). Reductions of tubulin and disruptions of microtubule structures induced by abnormal PrP mutants could be completely restored by overexpressing CK2α and CK2β in cells. Strong binding activity of the PrP mutants with CK2 will directly affect the interaction of CK2 with tubulin and other substrates (22).
-
The p21-activated kinases (PAKs) are a group of protein serine/threonine kinases; among them, PAK1 and PAK3 are highly expressed in brain tissues. Remarkable reductions of PAK3 and PAK1 were detected in scrapie-infected brains. The upstream activator Rac/cdc42 and downstream substrate Raf1, particularly the phosphorylated Raf1, were remarkably decreased at terminal stage, strongly highlighting correlation of dysfunction of Rac/cdc42-PAKs-Raf1 signaling with neuron loss during prion infection (23).
Wnt/β-catenin signaling is critical for synaptic disassembly and neuron loss. In prion infected rodent models, the brain levels of phosphor-β-catenin (Ser33,37and Thr41) were significantly up-regulated and its target substrate cyclin D1 was down-regulated. Increases of dickkopf-1, the antagonist of Wnt/β-catenin signaling, and decreases of Wnt-3 and phosphor-GSK3β were also found in scrapie-infected brains. Such abnormal changes displayed time-dependent progression along with the incubation period, reflecting a repression of Wnt/β-catenin signaling in neurons (24).
Polo-like kinases (PLKs) are a family with five major members. PLK3 phosphorylates Cdc25C on Ser216and inhibits the activity of Cdc25C, while PLK1 phosphorylates Cdc25C on Ser198and leads to activation and nuclear import, which is vital in synaptic plasticity. The increase of PLK1 and decrease of PLK3 were identified in scrapie-infected brains. Cdc25C and its phosphorylated forms were down-regulated, and Cyclin B1 and PCNA were obviously up-regulated. Aberrant changes of cell cycle regulatory PLKs-Cdc25C-Cyclin B1signaling in the prion-infected neurons lead to the cell cycle arrest at M phase (25).
Brain-derived neurotrophic factor (BDNF) appears primarily presynaptic effects at central, autonomic, and neuromuscular synapses. The brain level of BDNF was significantly reduced in scrapie-infected hamsters (26). The major components of BDNF signaling were also remarkably decreased, including tropomyosin-related kinase (Trk) receptors, phosphor-TrkB (p-TrkB), growth factor receptor-bound protein 2 (GRB2) and neurotrophin receptor p75 (p75NTR). Remarkable reduction of brain BDNF-TrK-GRB2-p75NTR signaling reflects a severe depletion of neurotrophic factors and a defect of energy metabolism in local microenvironment.
Nerve growth factor (NGF) is another member of neurotrophin family. A decrease of brain NGF was observed in the prion infected experimental animals and in prion infected cell line (27-28). Its upstream positive regulatory kinases, p-CREB, p-CaMKIV and CaMKK2, were decreased, whereas the negative regulatory one, p-CaMKK2, was increased. The aberrant situations of those kinases in prion infected cell lines could be also partially reversed by removal of prion agent by resveratrol. Prion infection affects the activity of CDK5-CaMKK2-CaMKIV-CREB cascade, which reduces the expression of NGF.
Glucose transporter 3 (GLUT3) mainly mediates the glucose passing through the membrane of neurons. The brain GLUT3 levels in scrapie-infected rodents and the prion infected cell line were significantly downregulated (29). The level of hypoxia-inducible factor-1 alpha (HIF-1α), which positively regulated the expressions of GLUTs, was also markedly downregulated in the scrapie-infected brains. The glucose uptake activity in the prion infected cell line was markedly decreased. It indicates a severe defect in glucose uptake and metabolism of neurons during prion infection.
-
Abnormal mitochondrial, oxidative and ER related stresses were observed in the prion infected brains and the cultured cells accumulated with PrPScor PrP mutants, involving numerous brain proteins and pathways (Figure 2). Reliable molecular interaction between PrP and 14-3-3βin vitroandin vivowas illustrated. Wild-type PrP was promoted, but PrP106-126 peptide inhibited the dimerization of 14-3-3β (30). Time-dependent reduction of 14-3-3 was detected in the brains of scrapie-infected mice, accompanied with the increase of itsS-nitrosylated form and many mitochondrial apoptotic agents. Challenge of PrP106-126 onto the cultured cells decreased the level of 14-3-3 and induced translocations of cellular Bax to the membrane fractions. Knockdown of 14-3-3 aggravated the mitochondrial apoptosis in PrP106-126 exposed cells (31).
 Figure 2.
Figure 2.Schema of prion-induced mitochondrial stress, ER stress, and oxidative stress in brains.
Note: Accumulations of PrPScand PrP mutants cause aberrant alterations (either expressing level or activity or translocation) of numerous brain proteins and pathways related to mitochondrial stress, ER stress, and oxidative stress.
Abbreviation: PrPSc=scrapie-like prion protein; PrP=prion protein; ER=endoplasmic reticulum.
Increases of ER-stress-related proteins Grp94, XBP1, TRAF2, and CHOP in the cells expressing PrP mutants with extra ORs (PG9 and PG12), while decrease of Bcl-2 and increase of cytochromeCin the cells expressing the mutant removal of ORs (PG0). The mutants with inserted ORs underwent mainly via ER stress and the mutant without OR via mitochondrial-related pathway (32). Accumulation of Cyto-PrP in cytoplasm also induces cell apoptosis, in which mitochondrial apoptotic pathway seems to be essential (33). Dynamin-related protein 1 (Drpl) and optic atrophy protein 1 (Opa1) are two essential elements for mitochondria fission and fusion. Reductions of brain Drp1 and Opa1 were noticed in various scrapie-infected animals, indicating a disturbance of brain mitochondria dynamics (34).
Swelling mitochondria structures were observed in the prion-infected cell line by transmission electron microscopy. The levels of Pink1 and Parkin, particularly in the mitochondrial fraction, were increased, whereas the levels of mitochondrial membrane proteins TIMM44, TOMM20, and TIMM23 were decreased. The brain Pink1 and Parkin in mice infected with scrapie were also increased at the terminal stage, predominantly located at the areas with more PrPScdeposit. Enhanced Pink-Parkin pathway mediated activated mitophagy during prion infection (35).
Cells expressing PrP mutants (KDEL and 3AV) produced an 18-kD large C-terminal proteolytic resistant fragment (Ctm-PrP). Those cells were more sensitive to ER stress stimuli, showing ER-mediated apoptosis by CHOP and the caspase-12 apoptosis pathway. Ctm-PrP and ER stress were only observed in the cells expressing PrP mutants in the transmembrane region (G114V and A117V), but not in those expressing the mutants outside the transmembrane region (36). Protein disulfide isomerase (PDI) is an enzymatic chaperone for reconstructing misfolded protein in ER lumen. In the scrapie-infected hamsters, abnormally upregulated brain PDI, Grp78 and Grp58 were detected. Expressions of PrP mutants in the cultured cells induced high levels of PDI and Grp18 and cytotoxicity. PDI and its relevant executors may function as a pleiotropic regulator in the processes of misfolded PrP proteins (37).
The OR domain was critical for PrP antioxidation. Disruption of ORs induced a high level of ROS and a low level of glutathione peroxidase in cells that were more susceptible to the challenges of oxidative factors (38). Abnormal increases of ROS are repeatedly reported in the brains of human and animal prion diseases. Molecular interaction between PrP and HS-1 associated protein X-1 (HAX-1), a protein associated with cell apoptosis. Co-expression of wild-type PrP and HAX-1 increased the cultured cells to resist the challenge of H2O2(39).
-
Although the clinical and general neuropathological features of prion disease have been well documented for a hundred years, the molecular abnormalities started to be revealed in the past three decades. Along with the development and progress of molecular biology and omics, the mysterious veil of the pathogenesis of CJD-associated neuropathological changes has been gradually revealed by uncovering the aberrant alterations of dozens of bioactive components and biological pathways. Extensive dysfunctions of numerous biological processes and signals directly lead to slow but irreversible damage on neurons and brain tissues infected with prions, the majority of them occurring much earlier than the appearance of clinical symptoms, even earlier than the appearance of detectable PrPSc. Other neurodegenerative diseases, such as AD and PD, also display similar neuropathological processes. However, besides the unique infectivity of prions, the clinical duration of PrD is much shorter, which possibly reflects a much faster and more severe neuropathological deterioration.
-
All staffs, doctoral and master students in the Department of Prion Disease of National Institute for Viral Disease Control and Prevention, China CDC since 1998 and the colleagues from provincial CDCs and sentinel hospitals in National Surveillance for prion diseases since 2006.
HTML
| Citation: |

|