
 Download:
Download:




-
Zoonotic diseases are increasing globally and pose a great threat to both livestock and human health. Zoonoses are estimated to cause over one billion cases of infection and millions of deaths annually worldwide. Over 30 new human pathogens have been detected in the last 3 decades with over 75% of these originating from animals (1). Establishment of a robust, integrated surveillance system with strong laboratory investigation abilities is urgently needed to eliminate zoonoses and minimize the risk of entry, establishment, and spread of emerging zoonoses (2).
Metagenomic next-generation sequencing (mNGS) can detect various pathogenic microorganisms without bias. mNGS is increasingly used to diagnose infectious diseases, particularly when conventional diagnostic approaches are limited. One example involved the use of mNGS for a chronic neuroleptospirosis case: traditional methods failed to identify the pathogen; however, mNGS identified the sequence asLeptospira santarosai(3). mNGS has been applied for clinical diagnosis and pathogen discovery in wildlife, especially for rare, novel, and difficult-to-detect pathogens. Rapid pathogen screening facilitates identification to initiate timely interventions.
The importance of zoonoses and their public health effects are well recognized (2). Knowledge of zoonotic pathogens in wildlife hosts or vectors in frequent contact with humans and domestic animals is important for understanding pathogenic diversity in nature (4). Metagenomic sequencing has unprecedented value in these investigations because it can describe microbial communities in a relatively unbiased manner, as opposed to screening for specific taxa (5). In wildlife investigations for zoonotic diseases, the targeted pathogens are typically predetermined, and samples are collected in the field which are transferred to the laboratory. Some issues with these surveys include that they only test for predetermined pathogens, the survey samples must be preserved and transferred under biosafety protocol, and the procedure is time-consuming. Long field deployments in geographically isolated places where entire laboratory teams must be sent each sentinel site are not only costly, but also increase stress on laboratory employees.
During the coronavirus disease 2019 (COVID-19) pandemic, the mobile polymerase chain reactions (PCR) laboratories provided a rapid, accurate, and clinically effective option for hospital diagnostic capabilities. In an East African Community (including Kenya, Rwanda, Burundi, Uganda and South Sudan), the mobile laboratory network for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) diagnostics ensured trade was able to continue during border closures (6). Here, during a mobile laboratory survey for rodents carryingYersinia pestisin the field, we performed mNGS onsite to detect possible pathogens in the rodent samples. We had planned to use the rodent samples forY. pestis-associated plague monitoring, butBartonellasequences were unexpectedly identified. With the sampling and sequencing performed onsite, mNGS provided a rapid and broad-spectrum survey model for detecting zoonotic pathogens in rodents.
-
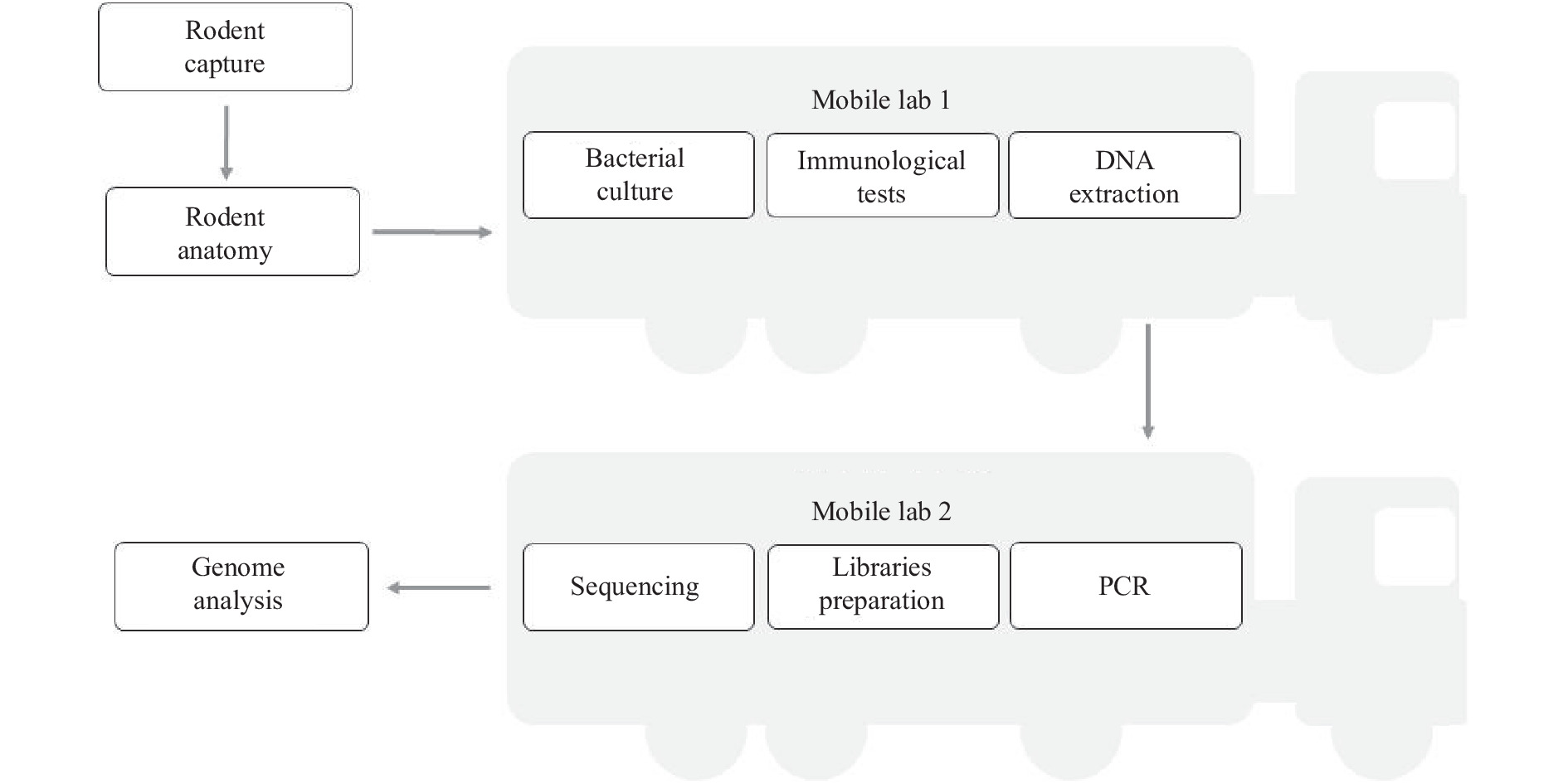
In September 2021, we conducted a field investigation for rodents carryingY. pestisin the Inner Mongolia Autonomous Region. The rodents were sampled using rat clips. The vehicle containing the mobile laboratory arrived at the surveillance sentinel so laboratory testing could be carried out, such as performing mNGS using a sequencer in the mobile laboratory (Figure 1). Heart, liver, lung, and serum samples were taken from the rodents.
 Figure 1.
Figure 1.Laboratory procedure from rodent capture to pathogen genome identification in the field monitoring based on the mobile laboratories.
Abbreviations: DNA=deoxyribonucleic acid; PCR=polymerase chain reactions. -
The diagnostic methods included identifyingY. pestisfrom isolates with a culture by a special phage lysis assay. Immunological methods for detecting the F1 antigen, i.e., the reverse indirect hemagglutination assay (RIHA) and/or colloidal gold-immunochromatography assay, were performed on liver and lung samples. Serum samples collected from the rodents were tested for antibodies against the F1 antigen via an indirect hemagglutination assay (IHA).
-
Total genomic DNA was extracted from the rodent hearts, livers and lungs using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). All genomic DNA samples were tested for caf1 and Chr392 via real-time PCR to detectY. pestis. ThessrAgene was tested using real-time PCR to detectBartonellaspp.(7).
-
DNA was quantified using Qubit v3. The sequencing libraries were constructed with the MGIEasy Fast PCR-FREE FS Library Prep Set (MGI, Wuhan, China), DNBSEQ OneStep DNB Make Reagent Kit V2.0 (OS-DB; MGI), Nextera XT DNA Sample Prep Kit (Illumina, California, USA) and Nextera XT Index Kit (Illumina). These libraries were sequenced on the portable DNBSEQ sequencing platforms (MGI) using a portable DNBSEQ Sequencing Set (SE100; MGI) and on the iSeq100 sequencing platforms (Illumina) using iSeq 100i1 Reagent (PE150; Illumina).
-
The mNGS data were processed using Kraken (8), the MGI Pathogeny Fast Identification pipeline, and the MicroFuture multiple pathogen analysis for metagenomic identification of pathogens. Bowtie2 and samtools were used to extract the pathogen-related reads.
To determine theBartonellaspecies from the pathogen-related reads, the extracted reads were blasted against the NCBI refseq database, the NR database, and theBartonellagenus genome sequences. ABartonellagenus genome reference database was constructed by retrieving all theBartonellagenera with genome sequences in the NCBI Assemble database. The sequences were evaluated using the average nucleotide identity (ANI) to filter obviously abnormal genomes. Finally, 144 whole-genome sequences belonging to 39 species were detected and included in the reference database. The extracted reads were then blasted against the reference database. The BLASTN cut-offs for the E-value was 0.00001 and the identity was 80%.
-
The mobile laboratories were comprised of one BSL-3 laboratory for sample preparation and pathogen culture along with one BSL-1 laboratory for PCR and sequencing. When CLOSS was performed in the field, the power supply vehicle and camping vehicle will be included. Three to four technicians working in the laboratories and two technicians for operation and maintenance of the mobile laboratories and power supply vehicle were needed.
The mobile laboratories arrived at the field site where rodent samples were collected. The electrical supply allowed us to use a refrigerator to store reagents. In the BSL-3 mobile laboratory, tissue was taken and DNA was extracted. In the BSL-1 mobile laboratory, the library was prepared and sequencing was initiated. The field-generated sequence data were analyzed using a variety of commercial and custom-developed bioinformatic workflows. The whole turnaround time of our protocol was less than 24 hours.
-
Total DNA was extracted from two rodent samples and tested forY. pestisusing real-time PCR, and all samples were negative. Additionally, all rodent samples used to detect the F1 antigen via colloidal gold immunochromatography assay and RIHA and the F1 antibody using IHA were negative.
-
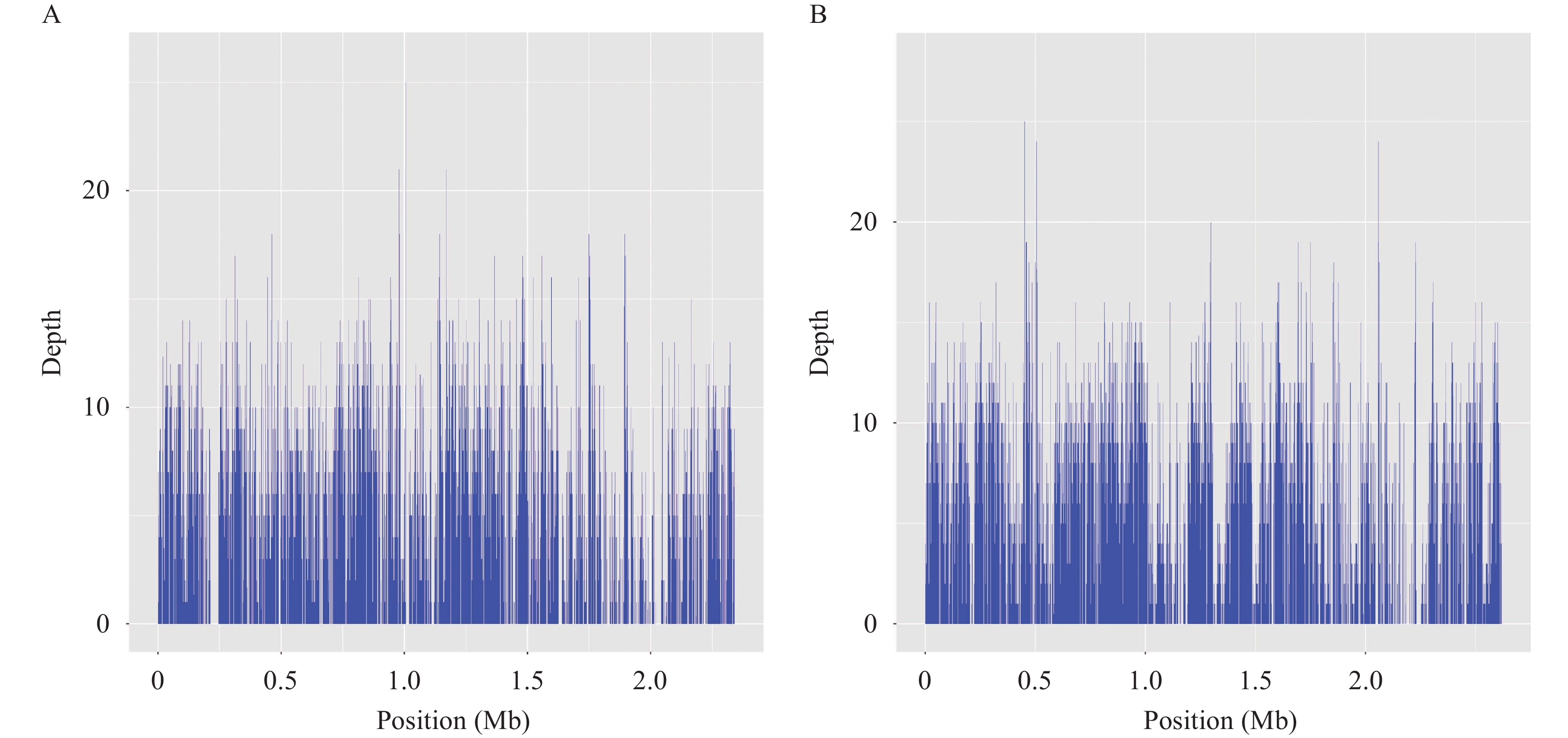
Metagenomic sequencing was performed on samples 11 and 12, which were identified asMeriones unguiculatus(gerbil) andSpermophilus dauricus(citellus). Sample 11 was the liver of aMeriones unguiculatus, which yielded about 1.4 Gb of sequencing data (5,017,499 reads, PE150) after 18 hours of sequencing. After filtering the quality reads and host reads, we obtained a dataset of 471,040 clean reads. As per the refseq database, 4,351 reads (including 253 unique reads) corresponded toBartonella grahamii(relative abundance: 49.26%) and 2,722 reads (including 70 unique reads) corresponded toB. tribocorum(relative abundance: 30.82%). The reads were then mapped to the reference genomes ofB. grahamii(NC_012846.1) andB. tribocorum(NC_010161.1), yielding approximately 22.4% and 7.8% coverages, respectively (Figure 2).
 Figure 2.
Figure 2.Mapping of genome assemblies forB. grahamii(A) andB. tribocorum(B) generated from metagenomic next-generation sequencing (mNGS) reads to the respective reference genomes.
Note: the reference genomes: B. grahamiiNC_012846.1 and B. tribocorumNC_010161.1.Virulence-associated genes such asomp89andomp43(coverage >70%) were identified as major putative Fn-binding proteins in theB. henselaeouter membrane proteins (9). Compared to the NR database, all 323 unique reads extracted from the refseq database corresponded toBartonellaspp. TheBartonellagenus database constructed comprised 144 whole-genome sequences belonging to 39 species. ANI analysis showed intraspecies nucleotide similarity rates of >97% (different strains within the same species;Supplementary Figure S1). Finally, from the 323 unique reads, 22 were species-specific, including 21 corresponding toB. grahamiiand one corresponding toB. tribocorum.
For sample 12, extracted from the liver of theSpermophilus dauricus, we obtained ~0.7 Gb of sequencing data (6,597,208 reads, SE100) after 9 hours of sequencing. After filtering the quality reads and the host reads, metagenomic sequencing yielded a dataset of 6,526,977 clean reads, of which only 29 corresponded toBartonellaspp. (relative abundance: 1.1%). We also obtained 2,673 reads related toAspergillus parasiticus(relative abundance near 99%) in the sequencing data from this sample.
-
We further detected theBartonellafrom samples 11 and 12 using real-time PCR. Both samples were positive forssrA, and the CT values were 27.24 for sample 11 and 32.88 for sample 12, indicating the correspondence of the relative abundances of the sequencedBartonellareads from both samples.
-
Animal-based surveillance can identify zoonotic pathogens and provide public health authorities with sufficient warning to implement appropriate prevention measures before human cases occur (10). mNGS is an unbiased, timely, and accurate approach to pathogen detection (11-12). Cultivating pathogens can take several days; mNGS can reduce the time needed to obtain results to less than 24 h (13). In this study, we identifiedBartonellaspp. from rodent samples.Bartonellaspp. are highly adaptable and can infect various hosts and vectors, with at least 13 species known to be pathogenic to humans (14). ManyBartonella-associated illnesses are linked with arthropod or animal transmission, and most patients reported having animal contact prior to disease onset. In Inner Mongolia, 4Bartonellaspecies were detected from 188 small mammals sampled from 2012–2013, and the positive infection rates ranged from 38.37%–56.41% (15). Here, we identifiedBartonellainfections from rodent tissue samples. We detected only two samples via mNGS, and both were positive. In this study, species-specific reads corresponding to twoBartonellaspecies were found inMeriones unguiculatus, suggesting possible complexBartonellaspp. infections in this rodent. Whether allBartonellaspp. were etiologic agents of human illness was unclear owing to the lack of accurate diagnostic methods in clinics, which were important for correct diagnoses and estimating disease burdens (16).Bartonellaprevalence and transmission remain unknown in China. Previous data and our findings demonstrate the necessity of surveillance of wild animals and people who are exposed to wild animals and their surroundings that have tested positive for Bartonella.
In the present zoonosis surveillance system, transferring animal carcasses from sampling locations in the field to the laboratory is time-consuming and requires biosafety protection. Zoonotic pathogen surveillance, such as plague monitoring in grasslands and plateaus, covers vast areas; thus, transferring samples may lower working efficiency. Mobile laboratory-based surveys save time and expand regional monitoring more effectively than previous strategies, which may include field sampling. Since mobile laboratory technologies are frequently designed to be durable, affordable, and mostly independent of energy, Internet access, or cold chain availability, they provide an excellent opportunity for local capacity building.
Additionally, mNGS can expand the monitored pathogen spectrum and is more advantageous for discovering new pathogens, especially for accurate laboratory diagnoses in regions without access to sophisticated laboratory techniques. We have named this zoonotic pathogen survey model “co-localization of sampling and sequencing (CLOSS),” which means performing animal-borne pathogen surveys in the field via metagenomic sequencing techniques in mobile laboratories. For monitoring zoonotic pathogen, CLOSS should be integrated into the Chinese pathogen identification net (China PIN). Technical laboratory staff should be trained in sequencing by China PIN. The CLOSS procedure will be further improved and practiced in order to evaluate and optimize this monitoring model. Due to COVID-19 outbreaks, many CDCs in China are now equipped with mobile laboratories. Our CLOSS model can expand the applications of these mobile laboratories to surveillance of other infectious diseases following the COVID-19 pandemic.
-
No conflicts of interest.
HTML
Sampling and Tissue Collection at Surveillance Sites
Bacterial Cultures and Identification
Genomic DNA Extraction and Real-Time PCR
Metagenomic Sequencing
Bioinformatics
Real-Time PCR and Serological Detection ofY. pestis
Metagenomic Sequencing
PCR Detection forBartonella
| Citation: |

|